|
P53
| P53
|
|
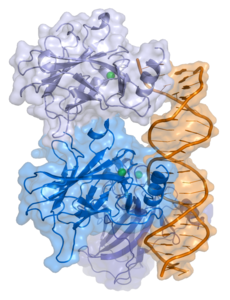
| Bänder-/Oberflächenmodell von p53 an DNA nach PDB 1TUP
|
|
Vorhandene Strukturdaten: 1a1u, 1aie, 1c26, 1gzh, 1hs5, 1kzy, 1olg, 1olh, 1pes, 1pet, 1sae, 1saf, 1sag, 1sah, 1sai, 1saj, 1sak, 1sal, 1tsr, 1tup, 1uol, 1ycs, 2ac0, 2ady, 2ahi, 2ata, 2b3g, 2bim, 2bin, 2bio, 2bip, 2biq, 2fej, 2gs0, 2h1l, 2j1w, 2j1x, 2j1y, 2j1z, 2j20, 2j21, 2ocj, 3sak
|
| Eigenschaften des menschlichen Proteins
|
| Masse/Länge Primärstruktur
|
393 Aminosäuren
|
| Kofaktor
|
Zn2+
|
| Isoformen
|
2
|
| Bezeichner
|
| Gen-Namen
|
TP53 ; LFS1; TRP53; p53
|
| Externe IDs
|
|
| Vorkommen
|
| Homologie-Familie
|
TP53
|
| Übergeordnetes Taxon
|
Wirbeltiere
|
Das Protein p53 ist in vielen Typen von entarteten Zellen in erhöhter Menge messbar. Es ist auch in normal wachsenden Zellen vorhanden. In ruhenden Zellen ist p53 jedoch kaum oder gar nicht zu finden. In vielen Tumortypen ist das für p53 kodierende Gen mutiert. In manchen, aber nicht allen Tumoren, scheint das Protein als Tumorsuppressor zu wirken. p53 spielt eine Rolle bei der Regulation des Zellzyklus, wo es die Aktivität einer Anzahl von Genen bremst. Zudem ist es in allen Wirbeltieren zu finden.[1]
p53 wurde 1979 unabhängig voneinander von Albert B. DeLeo, David P. Lane und Arnold Levine entdeckt.[2][3] Der humane Tumorsuppressor p53 reguliert als Transkriptionsfaktor nach DNA-Schädigung die Expression von Genen, die an der Kontrolle des Zellzyklus, an der Induktion der Apoptose (des programmierten Zelltods) oder an der DNA-Reparatur beteiligt sind. Aufgrund dieser Eigenschaft wird p53 in der Literatur als „Wächter des Genoms“ bezeichnet.[4] Die besondere medizinische Bedeutung erklärt sich aus dem Befund, dass p53 in 50 % aller menschlichen Tumoren mutiert ist. Der Verlust der p53-Funktion spielt daher eine kritische Rolle bei der Entstehung von Krebs, ist jedoch nicht dessen ursächlicher Auslöser. p53 wurde aufgrund seiner Bedeutung 1993 zum „Molekül des Jahres“ gewählt.
Funktionen von p53
p53 erhielt seinen Namen aufgrund der scheinbaren Molekularmasse von 53 kDa auf einem SDS-PAGE-Gel.
Das dazugehörige Gen, das TP53-Tumorsuppressor-Gen, liegt auf dem Chromosom 17p13.1. Um es von dem Protein zu unterscheiden, wird es kursiv geschrieben (TP53 ist die offizielle Bezeichnung für menschliches p53-Protein). Das p53-Protein ist von Natur aus instabil, wird aber regelmäßig „nachgebildet“. Kommt es in der DNA jedoch zu Schäden, etwa einem Doppelstrangbruch, die eine Replikation oder eine Mitose fehlerhaft ablaufen lassen würden, so wird p53 innerhalb von 30 Minuten posttranslational stabilisiert, wodurch sich p53 in der Zelle akkumuliert.
Die Akkumulation von p53 hat viele Folgen. So werden einerseits DNA-Reparatur-Mechanismen in Gang gesetzt, andererseits wird der Zellzyklus gestoppt. Letzteres geschieht dadurch, dass p53 als Transkriptionsfaktor die Produktion des Proteins p21 induziert. p21 wiederum hemmt sowohl den Cyclin D/CDK4/6-Komplex als auch den Cyclin E/CDK2-Komplex. Diese werden eigentlich benötigt, um einen vom Protein pRB gebundenen weiteren Transkriptionsfaktor (E2F) freizusetzen, der den Zellzyklus weiterführen würde. Die Zelle bekommt also durch p53 Zeit, sich selbst zu reparieren, bevor sie sich teilt. Ist die DNA wieder in Ordnung, so sinkt der p53-Spiegel wieder, p21 wird nicht mehr transkribiert und nach einer Weile geht der Zellzyklus weiter.
Wird p53 jedoch zu stark angehäuft und kommen weitere Faktoren hinzu, so aktiviert p53 Gene der Bcl2-Familie (insbesondere den Apoptose-Regulator BAX), die wiederum in Form einer Signalkaskade Caspasen auslösen und so zur Apoptose (programmierter Zelltod) führen. Demnach ist p53 wie eine Art Bremse, die nötig ist, um Zellen vom unkontrollierten Wachstum und weiterer Schädigung abzuhalten, und deren Mutation zu verstärkter Zellteilung führt.
Eine andere Funktion von p53 ist, dass es offensichtlich das menschliche Schwangerschaftshormon hCG steuert, wie Wissenschaftler der Universität Leipzig nachweisen konnten.[5]
Schädigung von p53
p53 ist ein Schlüsselprotein mit enormer Bedeutung. Ein Defekt hat demnach große Schadwirkung. Nach heutigem Kenntnisstand sind es im Allgemeinen Punktmutationen, die zu einem Funktionsverlust führen. Als Folge dieses Verlustes ist weder ein Anhalten des Zellzyklus zur DNA-Reparatur noch die Einleitung der Apoptose möglich. Die Zellen beginnen sich auch mit Schäden in der DNA unkontrolliert zu teilen, es kommt zur Tumorbildung. Ferner führt der Verlust des funktionsfähigen p53 zu einem Verlust der Synthesefähigkeit von Cytochrom-C-Oxidase 2. Dadurch kann die Cytochrom-C-Oxidase-2 Untereinheit nicht mehr in den Cytochrom-C-Oxidase-Proteinkomplex (Komplex IV der Atmungskette) eingebaut werden. Die Krebszelle verliert zwar den Stoffwechsel der aeroben Atmung, geht aber zur Energiegewinnung in die anaerobe Glykolyse über.[6]
Im Folgenden eine Liste der Krankheiten, die ausschließlich oder hauptsächlich auf Mutationen in TP53 (dem p53-Gen) zurückzuführen sind.
Beim Glioblastoma multiforme, WHO °IV, ist das p53-Gen ebenfalls mutiert: beim primären GBM <30 %, beim sekundären (durch Progression eines niedergradigen Glioms wie des diffusen Astrozytoms WHO °II oder des anaplastischen Astrozytoms WHO °III) GBM >65 % sowie beim Riesenzellglioblastom 30 bis 40 %.[9]
Patienten, die mit dem Li-Fraumeni-Syndrom geboren werden, haben eine angeborene Mutation in TP53. So kommt es bei Menschen mit dieser Mutation schon in frühester Kindheit zu diversen Tumoren, wie Brustkrebs, Leukämie, Hirntumoren und vieles mehr. Ursache für den Krebs ist jedoch vermutlich nicht die Mutation von TP53 selbst, sondern die hohe Zellteilungsrate während des embryonalen Wachstums, trotz Defekten in der DNA, sodass Schäden sich akkumulieren und weitere Gene für die Regulierung des Zellwachstums geschädigt werden können. Wie Heidelberger Forscher im Jahre 2012 in der Fachzeitschrift Cell beschrieben, weisen Tumoren in Patienten mit angeborenen TP53-Mutationen darüber hinaus gehäuft Merkmale von katastrophalen Chromosomenumlagerungen (Chromothripsis) auf. TP53-Mutationen könnten entweder Auslöser dieser massiven Erbgutschäden sein oder den Zelltod mittels Apoptose, trotz massiver Zerstörung der Chromosomenstruktur, verhindern.[10] Da jede Röntgenuntersuchung oder Chemotherapie die Mutationsrate erhöht, ist sowohl die Diagnose, als auch die Behandlung von Patienten mit Li-Fraumeni-Syndrom besonders schwierig.
Neben spontan auftretenden Mutationen gibt es auch andere Ursachen für Schäden an p53 beziehungsweise seinen Funktionen. So gibt es tumorinduzierende Viren (sogenannte Onkoviren), die p53 hemmen, abbauen oder dessen natürlichen Abbau beschleunigen. Diese Strategie nutzen die Viren, da auch virale Erkrankungen eine Apoptose auslösen können und somit die Viren an ihrer Weiterverbreitung hindern würden.
Weiterhin kann p53 durch chemische Stoffe geschädigt werden, zum Beispiel durch das im Tabakrauch enthaltene Benzo[a]pyren oder durch Aflatoxin. Diese Stoffe hinterlassen charakteristische Merkmale in der geschädigten DNA und können dadurch als Verursacher identifiziert werden.
Zusammenhang zwischen p53 und Lebenserwartung
Versuche an Fruchtfliegen deuten darauf hin, dass eine künstlich reduzierte Aktivität des Anti-Tumor-Proteins sich bei den Versuchstieren auf die Lebensdauer positiv auswirkt.[11] Verhält sich das Protein jedoch aktiver als normal (überaktiv), so altern die behandelten Fruchtfliegen wesentlich schneller als gewöhnlich.[12] Bei Mäusen wurde der gleiche Sachverhalt beobachtet.[13] Der Mechanismus selbst ist jedoch bislang ungeklärt.
Bei der (häufig schnellen) Bildung von Geweihen und Hörnern von Hornträgern verhindert p53 überschießendes Wachstum; zugleich haben die Tiere im Vergleich zu anderen Säugetieren eine fünffach niedrige Krebserkrankungsrate.[14]
Siehe auch
Literatur
- H. Kessler, J. Buchner u. a.: p53 – ein natürlicher Krebskiller: Einsichten in die Struktur und Therapiekonzepte. In: Angewandte Chemie. Band 118, 2006, S. 6590–6611. doi:10.1002/ange.200600611
- Pierre Hainaut, Klas Wiman (Hrsg.): 25 Years of p53 Research. Kluwer Academic Publishers, 2005, ISBN 1-4020-2920-9
- T. Stiewe: The p53 family in differentiation and tumorigenesis. In: Nature Reviews Cancer, Band 7, Nummer 3, März 2007, S. 165–168. doi:10.1038/nrc2072. PMID 17332760. (Review).
- G. Ferbeyre, S. W. Lowe: The price of tumour suppression? In: Nature, Band 415, Nummer 6867, Januar 2002, S. 26–27; doi:10.1038/415026a. PMID 11780097.
- M. Lacroix, R. A. Toillon, G. Leclercq: p53 and breast cancer, an update. In: Endocrine-Related Cancer, Band 13, Nummer 2, Juni 2006, S. 293–325; doi:10.1677/erc.1.01172. PMID 16728565. (Review).
- A. B. DeLeo, G. Jay, E. Appella, G. C. Dubois, L. W. Law, L. J. Old: Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. In: PNAS, Band 76, Nummer 5, Mai 1979, S. 2420–2424. PMID 221923. PMC 383613 (freier Volltext).
- Ingo B. Runnebaum: Das p53-Gen ist ein Tumorsuppressor und Zellzyklusregulator in Brustkrebszellen. Habilitationsschrift, Universität Ulm, 1997.
Weblinks
Einzelnachweise
- ↑ PROSITE documentation PDOC00301. Swiss Institute of Bioinformatics (SIB), abgerufen am 20. September 2011 (englisch).
- ↑ D. P. Lane, L. V. Crawford: T antigen is bound to a host protein in SV40-transformed cells. In: Nature, Band 278, Nummer 5701, März 1979, S. 261–263; PMID 218111.
- ↑ D. I. Linzer, A. J. Levine: Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. In: Cell, Band 17, Nummer 1, Mai 1979, S. 43–52; PMID 222475.
- ↑ D. P. Lane: Cancer. p53, guardian of the genome. In: Nature, Band 358, Nummer 6381, Juli 1992, S. 15–16; doi:10.1038/358015a0. PMID 1614522.
- ↑ Cell Cycle, 10(21), 3758–3767
- ↑ S. Matoba, J. G. Kang, W. D. Patino, A. Wragg, M. Boehm, O. Gavrilova, P. J. Hurley, F. Bunz, P. M. Hwang: p53 regulates mitochondrial respiration. In: Science, Band 312, Nummer 5780, Juni 2006, S. 1650–1653; doi:10.1126/science.1126863, PMID 16728594.
- ↑ R. F. Boynton u. a.: Loss of heterozygosity involving the APC and MCC genetic loci occurs in the majority of human esophageal cancers. Proc. Nat. Acad. Sci. 89/-/1992. S. 3385–3388. PMID 1565631
- ↑ F. Chakrani u. a.: Mutations clustered in exon 5 of the p53 gene in primary nasopharyngeal carcinomas from southeastern Asia. Int. J. Cancer 61/-/1995. S. 316–320. PMID 7729941
- ↑ Riede, Werner, Schaefer: Allgemeine und spezielle Pathologie. 5. komplett überarbeitete Auflage. Thieme Verlag
- ↑ T. Rausch et al.: Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. In: Cell, 2012, 148, S. 59–71; PMID 22265402
- ↑ J. Bauer u. a. in Current Biology
- ↑ G. Ferbeyre, S. W. Lowe: Aging: The price of tumour suppression? In: Nature, 415, 2002, S. 26–27
- ↑ S. D. Tyner u. a.: p53 mutant mice that display early ageing-associated phenotypes. In: Nature, 415, 2002, S. 45–53. doi:10.1038/415045a PMID 11780111
- ↑ Y. Wang et al.: Genetic basis of ruminant headgear and rapid antler regeneration. In: Science, 2019, 364eaav6335; doi:10.1126/science.aav6335, PMID 31221830
|
|

