A Hajos–Parrish–Eder–Sauer–Wiechert-reakció a szerves kémiában a prolin nevű aminosavval katalizált aszimmetrikus aldol reakció.
Az 1970 körül felfedezett Hajos–Parrish katalitikus eljárásban prokirális szénatomot tartalmazó triketont (1), 0,3 mmol prolin katalizátor segítségével N,N-dimetilformamid (DMF) oldószerben sikerült egy biológiai körülmények között végrehajtott aszimmetriás gyűrűzárásban átalakítani optikailag aktív (a síkban poláros fény polarizációs síkját elforgató) (+)-(3aS,7aS)-3a,4,7,7a-tetrahidro-3a-hidroxi-7a-metil-1,5(6H)-indándionná (+)-2, (100% kitermelés, 93,4% enantiomerfelesleg (ee)).
Ennek az optikailag aktív biciklusos ketolnak a sztereokémiája röntgendiffrakciós vizsgálatok alapján megfelel a digitoxigenin CD-gyűrűje kristályszerkezetének. A (+)-2 ketol savas dehidratálása az optikailag aktív éndionhoz, (+)-(7a,S)-7,7a-dihidro-7a-metil-1,5(6H)-indándionhoz (+)-3 vezet.

Hajos és Parrish nemcsak a 6,5-gyűrűs biciklusos ketolt (+)-2, hanem annak anguláris etil homológját (+)-4 is előállították. A vegyület sztereokémiája röntgendiffrakciós vizsgálatok alapján cisz konfigurációt mutat, de az nem felel meg a digitoxigenin CD-gyűrű kristály szerkezetének. Ennek valószínűleg az etilcsoport és a C-4 és C-6 axiális hidrogének közti 1,3-diaxiális kölcsönhatás az oka. Ez a kölcsönhatás a 7a-etilcsoportot a hattagú gyűrűben ekvatoriális, a 3a-hidroxilcsoportot pedig axiális helyzetbe irányítja.[1][2]
Hajos és Parrish a (+)-2 homológját, a 6,6-gyűrűs biciklusos ketolt (-)-5 is előállította. Ennek prolinnal katalizált szintézisében fontos az oldószer helyes megválasztása: a reakció N,N-dimetilformamidban (DMF) a 6,6-gyűrűs biciklusos ketolt, dimetil-szulfoxidban (DMSO) viszont az optikailag aktív Wieland–Miescher-ketont adja, ami a (+)-3 éndion 6,6-gyűrűs homológja.[3]

Az optikailag aktív Wieland–Miescher-ketont természetesen a 6,6-gyűrűs biciklusos ketol (-)-5 dehidratálásával is elő lehet állítani.
Az eljárások részletes leírása ábrákkal az eredeti német szabadalomban és tudományos cikkben találhatók meg.[1][2]
Eder, Sauer és Wiechert nem biológiai körülmények között 47 mol% prolinnal 1M perklórsav jelenlétében acetonitrilben forralva közvetlenül a fent leírt éndiont (+)-3 állította elő. A Hajos, Parrish biciklusos ketolokat így természetesen nem tudták elkülöníteni.[4] Harminchét évvel később egy új csoport folytatta a Schering berlini kutatóintézetében Eder, Sauer és Wiechert munkáját. A Hajos-Parrish katalitikus eljárás megismétlésével az új csoportnak is sikerült az optikailag aktív 6,5-biciklusos ketolt előállítani.[5] A vegyület előállítását eddig csak a Hajos–Parrish közleményekben lehetett megtalálni.[1][2]
A kísérleti munka Hajos 1970 novemberében történt lemondása után hosszú ideig szünetelt ezen a szakterületen. 2000-ben Benjamin List és munkatársai azt találták, hogy intermolekuláris aldol addíciókat is lehetséges aldehidek és ketonok között létrehozni, csak lényegesen több prolint kell használni.[6]

A reakcióban nagy mennyiségű acetont (a keton reagens) kell használni, hogy a ketonnak prolinnal oxazolidinonhoz vezető reakcióját, valamint az aldehidnek prolinnal azometin-ilidhez vezető reakcióját visszaszorítsák.
List és munkatársai kiterjesztették a reakciót 1,2-diolok előállítására is.[7]
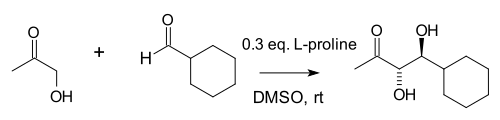
Számos vegyületet vizsgáltak meg egy program keretében. Prolin és 5,5-dimetil-tiazolidinium-4-karboxilát tiazóliumsó voltak a leghatékonyabb katalizátorok az aminok nagy csoportjában.[8]
A Wieland–Miescher-keton aszimmetriás szintézise egy másik intramolekuláris prolinnal katalizált reakció, amit Barbas és munkatársai 2000-ben újra megvizsgáltak.[9]
2002-ben David MacMillan és munkatársai kiterjesztették a prolinnal katalizált reakciót aldehideknek egymással történő aldol addíciójára.[10]
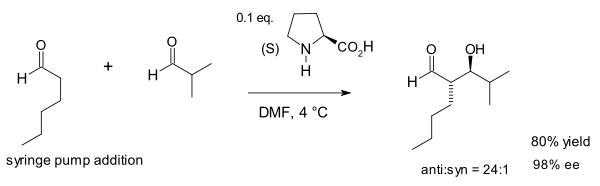
A reakció azért fontos, mert aldehidek rendszerint önmagukkal kondenzálnak.
Reakciómechanizmus
A triketon prolinnal katalizált aszimmetriás gyűrűzárására számos reakciómechanizmust javasoltak az évek során. Hajos (1974) egy enamin és egy hemiaminal (karbinolamin) mechanizmust is javasolt. Az enamin köztiterméket sztöchiometrikus mennyiségű 18O-dúsított vízzel végzett kísérletei alapján vetette el. E kísérletek ugyanis csak 7,2% 18O-dúsulást jeleztek. Egy ellenőrző kísérletben viszont maga a (+)-2 ketol reakciótermék 34,1% 18O-dúsulást mutatott. Az oxigénizotóp beépülését 18O-jelzett CO2 tömegspektrometriás mérésével határozták meg.[1][2]
A Hajos által 1974-ben javasolt hemiaminál (karbinolamin) egy iminium hidroxid tautomerré változhat. Az oldalláncban levő metil keton az iminium hidroxid hatására enolizál, majd gyűrű zárással az optikailag aktív biciklusos ketol reakció terméket (+)-2 adja a katalitikus mennyiségű (S)-(-)-prolin jelenlétében.
Hajos későbbi vizsgálódásai azt mutatták, hogy a Swaminathan és munkatársai által javasolt molekuláris sablon mechanizmus („template mechanism”) alacsonyabb energiaszinteket mutat, mint akár az enamin, akár a karbinolamin mechanizmus. A mechanizmus lényege, hogy a triketon prolinnal katalizált aszimmetriás gyűrűzárásában, az oldószerben gyakorlatilag oldhatatlan prolin molekuláris sablonként egy hidrogénkötéses komplexben fejti ki katalitikus és sztereo-dinamikus hatását.[11] Ez a mechanizmus nemcsak az intramolekuláris, hanem az intermolekuláris aldolreakciókat is megmagyarázhatja. Így nem kellene különleges elméletekkel magyarázni a víz jótékony hatását a köztudottan vízre érzékeny enamin köztitermékekre.
Az Agami-mechanizmus (1984) reakciókinetikai kísérletek alapján olyan enamin köztiterméket tételez fel, amelyben két prolin molekula vesz részt az átmeneti szerkezetben.[12] Houk mechanizmusa szerint (2001) egyetlen prolin molekula elegendő egy ciklusos átmeneti állapot létrehozására, amelyben a prolin karboxilcsoportja hidrogénkötésben van.[13][14]

List és munkatársai 2000-ben az intermolekuláris reakciókra is enamin köztiterméket tételeztek fel, és a megfigyelt sztereoszelektivitást a Zimmerman–Traxler-modell segítségével a Re oldalú (Re face) közelítéssel magyarázták. A prolin ketonnal képzett enaminja és az aldehid közti Re faciális közelítés azért kedvezőbb, mert a térigényes R csoport és az ugyancsak térigényes prolin egység a szék konformációjú, hattagú gyűrűs komplex ellentétes oldalán helyezkedik el. A komplexet a két oxigént és a prolin nitrogénjét összekötő hidrogénkötés tartja össze.
Az Si oldalú közelítésben a két térigényes csoport a szék konformációjú, hattagú gyűrűs komplex azonos oldalán helyezkedik el, térbeli taszítás áll elő, ezért a reagáló molekulák Si faciális közelítése előnytelen.[15] Mindezeket a következő ábra mutatja:
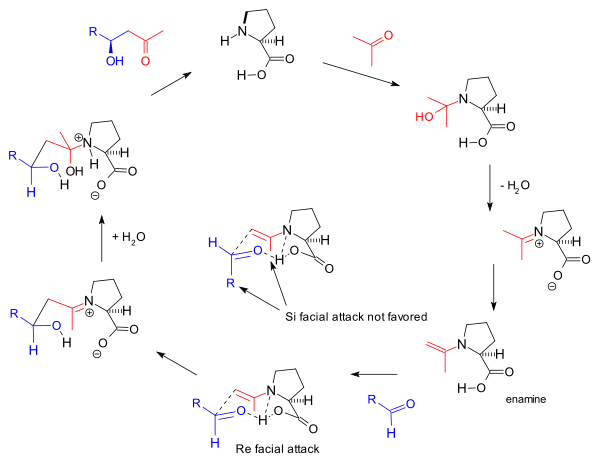
Houk és List szerint az eredeti Hajos-Parrish triketon reakciót is az enamin mechanizmus vezérli, de az Agami által javasolt két prolin molekulás köztiterméket[12] reakciókinetikai megfontolások alapján kétségbe vonták.[16] A reakció mechanizmust List H218O vízben végzett kísérlete támasztja alá, amely az oxigénizotópnak a reakciótermékbe való beépülését mutatja.[17] List ezért kizárta a Hajos féle aminal (karbinolamin) mechanizmust. List ugyanebben a cikkben leírta prolinnak acetonnal való reakcióját is, ami oxazolidinonhoz vezet:

Miután ennek a reakciónak az egyensúlyi állandója csak 0,12, ezért List arra a következtetésre jutott, hogy az oxazolidinon csak ún. „parasitic” (parazita-) szerepet játszik.
Blackmond 2004-ben oxazolidinon köztitermékeket talált propanal és nitrozo-benzol prolinnal katalizált alfa-aminooxilációjában (NMR-es meghatározás):[18]

Chiong Teck Wong a szingapúri High Performance Computing Intézetben ugyanezt az oxiaminációs reakciót vizsgálta nitrozo-benzol és n-butanal modellen optikailag aktív prolinol-szilil-éter katalizátorral.[19] Wong kutatási eredményei megerősítik, hogy a katalizátor egy enolt hoz létre, és egy enol-katalizátor komplexet képez. A nitrozo-benzol azután az enol-katalizátor komplexszel reagálva főtermékként az (S)-N-nitrozo aldol terméket hozza létre a Pauling-féle elektronegativitási táblázatnak megfelelően. Az aldol keverék nátriumborohidrides redukciója a megfelelő alkoholokhoz vezet, ahol a PN/PO arány ≥ 99:1. A Wong által javasolt enol képző mechanizmus jól magyarázza a kísérletileg megfigyelt nagyfokú enantioszelektivitást. Wong eredményeit az alábbi ábra mutatja.

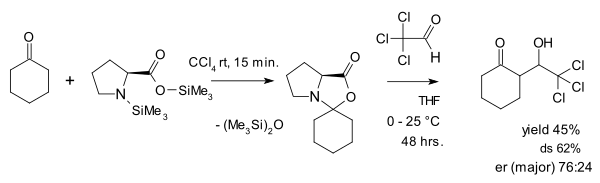
Seebach és Eschenmoser 2007-ben egy 47 oldalas (!) cikkben vitatták meg az oxazolidinoknak mint „parazita” köztitermékeknek a szerepét.[20] Cikkükben azt fejtegetik, hogy az oxazolidinok valójában kulcsponti szerepet játszanak a prolinnal katalizált reakciókban. Ennek alátámasztására ciklohexanonból és egy aktivált prolin származékból oxazolidinont állítottak elő, és azt klorállal reagáltatva aldol addíciós reakcióban a ciklohexanon és klorál aldolszármazékát nyerték 45%-os kitermeléssel. A diasztereo-szelektivitás (ds) 62%-os, az enantiomer arány (er) 76:24. A reakciót a következő ábra mutatja:
A reakció elnevezésének története
1985-ben Claude Agami professzor és munkatársai nevezték el először ezt a prolinnal katalizált Robinson típusú annulációt Hajos–Parrish-reakciónak.[21] 1986-ban Henri B. Kagan és Claude Agami professzorok[22] még mindig Hajos–Parrish-reakciónak nevezték a reakciót cikkük összefoglaló (Abstract) részében. 2001-ben Kagan professzor egy cikket közölt ”Nonlinear Effects in Asymmetric Catalysis: A Personal Account” címmel a Synlett c. folyóiratban.[23] E cikkben Kagan professzor már a „Hajos–Parrish–Wiechert-reakció” nevet használta. 2002-ben Benjamin List professzor még két nevet adott hozzá, és bevezette a „Hajos–Parrish–Eder–Sauer–Wiechert-reakció” elnevezést.[24] Az újabban szerves katalízisnek („Organocatalysis”) nevezett tudományos területen közölt cikkek még 2008-ban is 1985-ös, 2001-es, vagy 2002-es reakciónevet használnak.
Jegyzetek
- ↑ a b c d . Hajos, Z, G.; Parrish, D. R. Asymmetric Synthesis of Optically Active Polycyclic Organic Compunds. German Patent DE 2102623, July 29, 1971.
- ↑ a b c d Asymmetric synthesis of bicyclic intermediates of natural product chemistry Zoltan G. Hajos, David R. Parrish J. Org. Chem.; 1974; 39(12); 1615-1621. doi:10.1021/jo00925a003
- ↑ „Wieland–Miescher-keton” szócikk
- ↑ New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures Angewandte Chemie International Edition in English Volume 10, Issue 7, Date: July 1971, Pages: 496-497 Ulrich Eder, Gerhard Sauer, Rudolf Wiechert doi:10.1002/anie.197104961
- ↑ J.Org.Chem., 2008, 73, 5151-5154
- ↑ a b Proline-Catalyzed Direct Asymmetric Aldol Reactions Benjamin List*, Richard A. Lerner, and Carlos F. Barbas III J. Am. Chem. Soc. 2000; 122, 2395-2396 doi:10.1021/ja994280y
- ↑ Catalytic Asymmetric Synthesis of anti-1,2-Diols Wolfgang Notz and Benjamin List J. Am. Chem. Soc. 2000; 122(30) pp 7386 - 7387; (Communication) DOI: 10.1021/ja001460v
- ↑ Amino Acid Catalyzed Direct Asymmetric Aldol Reactions: A Bioorganic Approach to Catalytic Asymmetric Carbon-Carbon Bond-Forming Reactions Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C. F., III J. Am. Chem. Soc. (Article); 2001; 123(22); 5260-5267. doi:10.1021/ja010037z
- ↑ A proline-catalyzed asymmetric Robinson annulation reaction Tetrahedron Letters, Volume 41, Issue 36, September 2000, Pages 6951-6954 Tommy Bui and Carlos F. Barbas doi:10.1016/S0040-4039(00)01180-1
- ↑ The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes Alan B. Northrup and David W. C. MacMillan J. Am. Chem. SOC. 2002; 124, 6798-6799 doi:10.1021/ja0262378
- ↑ Proline-catalysed asymmetric ketol cyclizations: The template mechanism revisited R.Malathi, D. Rajagopal, Zoltan G. Hajos and S. Swaminathan, J.Chem. Sci., Vol. 116, No.3, May 2004, 159-162
- ↑ a b a b Stereochemistry-59 : New insights into the mechanism of the proline-catalyzed asymmetric robinson cyclization; structure of two intermediates. asymmetric dehydration Tetrahedron, Volume 40, Issue 6, 1984, Pages 1031-1038 Claude Agami, Franck Meynier, Catherine Puchot, Jean Guilhem and Claudine Pascard doi:doi:10.1016/S0040-4020(01)91242-6
- ↑ The Origin of Stereoselectivity in Proline-Catalyzed Intramolecular Aldol Reactions Bahmanyar, S.; Houk, K. N. J. Am. Chem. Soc. (Communication); 2001; 123(51); 12911-12912. doi:10.1021/ja011714s
- ↑ Transition States of Amine-Catalyzed Aldol Reactions Involving Enamine Intermediates: Theoretical Studies of Mechanism, Reactivity, and Stereoselectivity Bahmanyar, S.; Houk, K. N. J. Am. Chem. Soc. 2001; 123(45); 11273-11283 doi:10.1021/ja011403h
- ↑ a b Proline-Catalyzed Direct Asymmetric Aldol Reactions Benjamin List*, Richard A. Lerner, and Carlos F. Barbas III J. Am. Chem. Soc. 2000, 122, 2395-2396 doi:10.1021/ja994280y
- ↑ Kinetic and Stereochemical Evidence for the Involvement of Only One Proline Molecule in the Transition States of Proline-Catalyzed Intra- and Intermolecular Aldol Reactions Linh Hoang, S. Bahmanyar, K. N. Houk, and Benjamin List J. Am. Chem. Soc. 2003, 125, 16-17 doi:10.1021/ja028634o
- ↑ Asymmetric Catalysis Special Feature Part II: New mechanistic studies on the proline-catalyzed aldol reaction Benjamin List, Linh Hoang, and Harry J. Martin PNAS 2004 101: 5839-5842; doi:10.1073/pnas.0307979101
- ↑ Probing the Active Catalyst in Product-Accelerated Proline-Mediated Reactions Iwamura, H.; Wells, D. H., Jr.; Mathew, S. P.; Klussmann, M.; Armstrong, A.; Blackmond, D. G. J. Am. Chem. Soc. (Communication); 2004; 126(50); 16312-16313. doi:10.1021/ja0444177
- ↑ A theoretical investigation on the mechanism of the alpha,alpha-diphenylprolinol trimethylsilyl ether-catalyzed oxyamination reaction, Chiong Teck Wong, Tetrahedron Letters 50 (2009) 811-813.
- ↑ Are Oxazolidinones Really Unproductive, Parasitic Species in Proline Catalysis? - Thoughts and Experiments Pointing to an Alternative View Helvetica Chimica Acta Volume 90, Issue 3, Date: March 2007, Pages: 425-471 Dieter Seebach, Albert K. Beck, D. Michael Badine, Michael Limbach, Albert Eschenmoser, Adi M. Treasurywala, Reinhard Hobi, Walter Prikoszovich, Bernard Linder doi:10.1002/hlca.200790050
- ↑ A New Diagnostic Tool for Elucidating the Mechanism of Enantioselective Reactions. Application to the Hajos-Parrish Reaction, Agami, C.; Levisalles, J.; Puchot, C. J. Chem. Soc., Chem. Commun. 1985, 8, 441-442
- ↑ Nonlinear Effects in Asymmetric Synthesis C. Puchot, O. Samuel, E. Dunach, S. Zhao, C. Agami, H. B. Kagan, J. Am. Chem. Soc. 1986, 108, 2353-2357
- ↑ Nonlinear Effects in Asymmetric Catalysis: A Personal Account H.B. Kagan et al., Synlett 2001, 888–899
- ↑ Proline-catalyzed asymmetric reactions B. List, Tetrahedron 58 (2002) 5573-5590









2009-811.svg)

